Sekvenování nové generace (Next Generation Sequencing; NGS) je metoda vysoce výkonné analýzy primární struktury nukleových kyselin. Deset let od uvedení prvního komerčního NGS analyzátoru firmou 454 (nyní Roche) se aplikace NGS stávají rutinní součástí klinické praxe v řadě lékařských disciplín. Jednou z oblastí zaznamenávající významný pokrok díky možnostem NGS je i onkogenetika, která umožňuje identifikaci genetických faktorů zodpovědných za vysoké riziko vzniku zhoubných nádorů v postižených rodinách.
Sekvenování nové generace
Technologickým předpokladem nástupu NGS na počátku tisíciletí byl rychlý pokrok v oblasti výpočetní techniky a optoelektroniky, který umožnil spolehlivou detekci biochemických změn, ke kterým dochází při sekvenování velkého počtu (105–109) paralelně probíhajících sekvenačních reakcí. Technologická a biochemická podstata řešení NGS definuje jednotlivé přístupy (platformy), jejichž společným jmenovatelem je obrovský sekvenační výkon ve srovnání s „klasickým“ (Sangerovým) sekvenováním.1 Zatímco klasickými postupy je na jednom analyzátoru možné přečíst přibližně 105 bází genomové DNA denně, výkon NGS sekvenování je v závislosti na typu analyzátoru vyšší 103–107násobně. Velikost datového výstupu NGS určuje vysokou bioinformatickou náročnost vyžadující výkonné systémy pro zpracování získaných sekvenačních dat a znalost bioinformatických postupů pro jejich následnou analýzu. V reálném čase lze však nyní pomocí NGS získat kompletní genetickou informaci lidského genomu, a to s ekonomickými náklady představitelnými i pro českou laboratoř (v řádech desítek tisíc Kč). Pro srovnání: náklady na sekvenování lidského genomu v projektu Human Genome Project (1988–2003) činily 3,8 mld. USD; náklady na program Apollo (1961–1972) 25 mld. USD.2,3
Třebaže úvaha o dekódování personálního lidského genomu je atraktivní, její klinická využitelnost v lékařské praxi je zatím okrajová. Zásadní klinický přínos NGS spočívá v současnosti v tzv. cíleném sekvenování umožňujícím analýzu zvolených oblastí genomu (či transkriptomu). Výběr cílených oblastí umožňuje omezit celkovou velikost analyzované sekvence u vyšetřované osoby pouze na oblast zájmu a tím umožňuje využít vysoký výkon sekvenačního výstupu NGS k analýze několika (i desítek) pacientů v jednom sekvenačním běhu. V těchto modalitách je v současnosti cílené sekvenování vhodnou a ekonomicky únosnou technologií pro analýzy genetické predispozice k řadě vrozených onemocnění včetně dědičných nádorových syndromů.
Základem cíleného sekvenování je selektivní výběr sekvenačních templátů. V praxi se nejčastěji používá amplikonové NGS, kdy jsou cílové oblasti DNA obohaceny pomocí multiplexní PCR, nebo tzv. panelové sekvenování, při kterém dochází k vychytávání cílových oblastí z fragmentované DNA pomocí hybridizačních sond (tzv. sequence capture nebo sequence enrichment). Zatímco amplikonové sekvenování je vhodné spíše pro analýzy menšího počtu templátů, panelové sekvenování umožňuje selektivní vychytávání velkého počtu cílových oblastí (např. exonů stovek genů) až do celkové velikosti exomu (> 20 000 genů s >107 bp genetické informace). Oba postupy obohacení cílové DNA mohou být použity pro analýzy jednotlivých genů nebo jejich sad – genových panelů v podobě množství dostupných komerčních kitů. Je však možné je využít i pro vlastní návrh cílových oblastí NGS.
Pro panelové sekvenování templátů obohacených selektivním vychytáváním fragmentované genomové DNA existuje v současnosti několik přístupů, které se liší technologickým postupem, vhodností k použití v konkrétních projektech a v neposlední řadě i cenou. Mezi nejčastěji používané patří SeqCap EZ Choice (Nimblegen/Roche), Nextera (Illumina) a SureSelect či HaloPlex (Agilent). Všechny umožňují návrh vlastních sekvenačních panelů, které mohou být použity v diagnostice dědičných nádorových syndromů s využitím různých sekvenačních platforem. Výhodou systému SeqCap EZ Choice je dobrá homogenita pokrytí cílové sekvence, relativně malý podíl templátů, které nejsou ve skutečnosti cíleny k selektivnímu obohacení (off-target sekvencí), robustnost přípravy, ale i ekonomická rentabilita při realizaci velkého počtu analýz.4,5
Dědičné nádorové syndromy
Zhoubné nádory tvoří čtvrtinu všech úmrtí a představují druhou nejčastější příčinu smrti v České republice.6 Ve valné většině nádorová onemocnění vznikají v důsledku mnohaleté kumulace mutací genomové DNA v postižené tkáni.7 Přestože drtivá většina vznikajících mutací je v buňkách organismu účinně opravována vysoce efektivními mechanismy reparace genomové DNA, vzrůstá s rostoucím věkem pravděpodobnost, že některá z alterací zůstane neopravena, což dokládá i s věkem se zvyšující incidence nádorových onemocnění v populaci. Přeměnu normálních buněk tkání na nádorové buňky umožňují především aktivační mutace v jedné z alel protoonkogenů (způsobující aberantní syntézu jejich proteinových produktů se zvýšenou či změněnou funkcí) nebo inaktivace obou alel tumor supresorových či DNA reparačních genů (způsobující ztrátu funkce jimi kódovaných proteinů).
Většina nádorových onemocnění (> 90 %) vzniká jako tzv. „sporadické“ nádory, které se vyvíjejí na podkladě mutací vznikajících v průběhu pacientova života v buňkách postižené tkáně. Kromě této dominantní skupiny se nádorová onemocnění vyskytují i v podobě dědičných, tzv. „hereditárních“, nádorů, jejichž podstatou je nosičství patogenní mutace v některém z nádorových predispozičních genů.8 Většinu z doposud známých (> 150) nádorových predispozičních genů tvoří tumor supresorové geny lokalizované na autozomech. Jejich proteinové produkty slouží především jako negativní regulátory dělení či přežívání buněk nebo jako proteiny účastnící se oprav genomové DNA. Přítomnost vrozené mutace, která se u nosičů vyskytuje ve všech somatických buňkách, výrazně zvyšuje pravděpodobnost, že v průběhu života dojde v některé z buněk i k inaktivaci druhé alely genu způsobující funkční poruchu jeho genového produktu a iniciaci nádorového procesu.
Ve srovnání s normální populací jsou nosiči mutací v nádorových predispozičních genech ohroženi zvýšeným rizikem vzniku nádorového onemocnění, které se často vyskytuje v postižených rodinách opakovaně. Důvod, proč u nosičů mutací určitých genů vznikají nádorová onemocnění v typických lokalizacích, je mnohdy nejasný. U nosičů patogenních hereditárních mutací však onkologická onemocnění vznikají ve výrazně nižším věku a nemocní mají zvýšené riziko rekurence onemocnění. Ve většině případů je u nosiče mutace v některém z nádorových predispozičních genů přítomna jedna patogenní alela (mutace), a proto existuje na pohlaví nezávislé 50% riziko přenosu této mutace od jednoho z rodičů na jeho potomky. Zmíněné charakteristiky dědičných nádorových syndromů potvrzují důležitost diagnostiky přítomnosti patogenních mutací nádorových predispozičních genů u této skupiny nemocných a jsou racionálním důvodem pro jejich genetické testování. To je indikováno obvykle u onkologických pacientů s výskytem nádorového onemocnění v atypicky mladém věku (např. karcinom prsu ve věku do 40 let), u nemocných z rodin s mnohočetným výskytem charakteristických nádorových onemocnění (např. kolorektální karcinom u tří přímých příbuzných) nebo u nemocných se zhoubnými nádory s vybranými charakteristikami (např. tzv. „triple negativní“ karcinom prsu - histologický typ nádoru nevykazující expresi estrogenního, progesteronového ani HER2/Neu receptoru v nádorových buňkách. Výskyt této formy karcinomu prsu je zvýšen u nosiček mutací v genu BRCA1 (ale nikoliv BRCA2)). Pravděpodobnost přítomnosti patogenních mutací v nádorových predispozičních genech zvyšuje kombinace zmíněných charakteristik (např. mnohočetný výskyt triple negativních karcinomů prsu u osob v mladém věku a karcinomů ovaria v rodině).
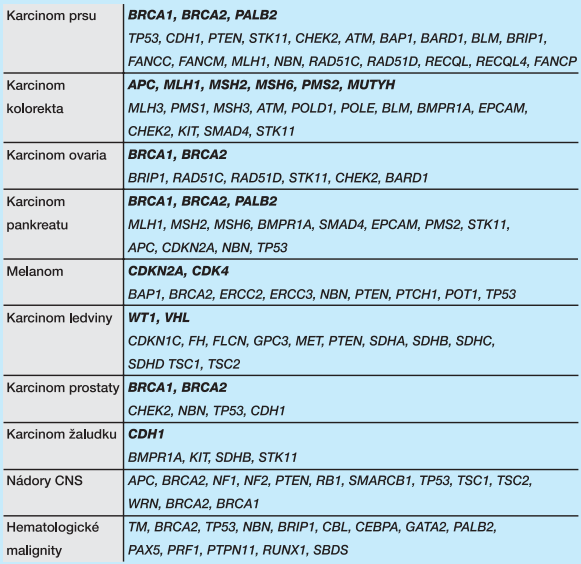
Indikaci ke genetickému vyšetření a interpretaci jeho výsledku provádí klinický genetik v součinnosti s molekulárními biology, onkology, gastroenterology, gynekology, dermatology a dalšími specialisty. Ti se podílejí na následném sledování nosiče patogenní mutace (a případně jeho příbuzných) z důvodu prevence vzniku (dalšího) nádorového onemocnění a na výběru optimální léčebné modality při výskytu onkologického onemocnění nebo indikaci doporučené chirurgické prevence (např. odstranění vaječníků či mléčné žlázy u nosiček mutací v genech BRCA1/BRCA2, nebo kolektomii u nosičů mutací v genu APC). U jednotlivých nádorových syndromů byly charakterizovány mutace v řadě predispozičních genů (tab. 1). Přitom pravděpodobnost výskytu patogenních alterací je často značně rozdílná. Např. zatímco pravděpodobnost mutací v genu BRCA1 či BRCA2 u pacientky s karcinomem prsu z rodiny se třemi dalšími příbuznými s karcinomem prsu je více než 50%, pravděpodobnost výskytu mutace v genu PALB2 v takovéto rodině nedosahuje 10 % a pravděpodobnost výskytu mutace v STK11 je hluboce pod 1 %.
Do nedávné doby byly jednotlivé nádorové predispoziční geny vyšetřovány u indikovaných pacientů postupně, obvykle v pořadí pravděpodobnosti výskytu patogenní mutace v populaci. Např. při výskytu dědičné formy karcinomu prsu byl v ČR nejprve analyzován gen BRCA1, jehož mutace jsou v naší populaci přibližně třikrát častější než mutace genu BRCA2. Protože je však mnoho predispozičních genů velmi rozsáhlých, jsou kompletní vyšetření jejich kódující sekvence klasickými postupy (doplněná o nezbytná vyšetření intragenových přestaveb) časově extrémně náročná. Z důvodu raritního výskytu patogenních mutací v řadě predispozičních genů se jejich nákladná klinicko-genetická vyšetření mohla provádět jen velmi omezeně a obvykle pouze v případech, kdy postižená rodina splňovala další kritéria naznačující přítomnost patogenní varianty konkrétního genu. Jejich rutinní analýzy by však z důvodů časové i finanční náročnosti nebyly odůvodnitelné. Při analýzách jednotlivých genů bylo možné nalézt přítomnost patogenní varianty přibližně u 20 % vyšetřovaných pacientů rekrutujících se z osob se zvýšenou pravděpodobností výskytu dědičných forem karcinomu prsu anebo ovarií.
Současný stav analýzy nádorové predispozice a jeho další perspektivy
Dostupnost NGS sekvenování umožňuje analýzu nádorových predispozičních genů především formou sekvenačních panelů. V panelovém sekvenování vzorků od pacientů s nádorovou predispozicí se setkáváme se dvěma mírně odlišnými přístupy. Zatímco některá pracoviště provádějí analýzu nádorové predispozice s použitím nádorově specifických sekvenačních panelů (obsahují obvykle několik až několik desítek cílených predispozičních genů), v dalších jsou používány komplexnější sekvenační panely obsahující kolekci známých predispozičních genů bez ohledu na nádorovou specifičnost (např. True-Sight Cancer Panel; 94 genů a 284 SNPs asociovaných s dědičnými nádory)9 nebo panely obohacené i o kandidátní geny, u nichž se předpokládá, že s nádorovou predispozicí souvisí (např. CZECANCA Panel; 219 genů).10
První přístup využívají obvykle laboratoře velkých zdravotnických komplexů nebo diagnostické společnosti analyzující nádorové predispoziční geny (např. Myriad Genetics, Ambry Genetics, GeneDx), které zpracovávající desítky až stovky vzorků denně. Výhodou tohoto přístupu je vysoká rentabilita a rychlost vyšetření. Pro podmínky laboratoří v ČR, které zpracovávají obvykle pouze několik set vzorků ročně, je však výhoda malých sekvenačních panelů diskutabilní a převažují výhody použití komplexnějších panelů. Mezi ně patří operativnější využití sekvenační kapacity a tím i kratší doba realizace vyšetření (v řádu týdnů). Zásadní výhodou komplexnějších nádorových panelů je však zjištění, že mutace v genech doposud asociovaných pouze či převážně s určitým typem nádorů mohou ovlivňovat riziko vzniku nádorových onemocnění i v dalších lokalizacích (např. mutace v mismatch repair genech podmiňující hereditární nepolypózní kolorektální karcinom zvyšují riziko vzniku karcinomu endometria, ale pravděpodobně i například karcinomu prsu).11,12
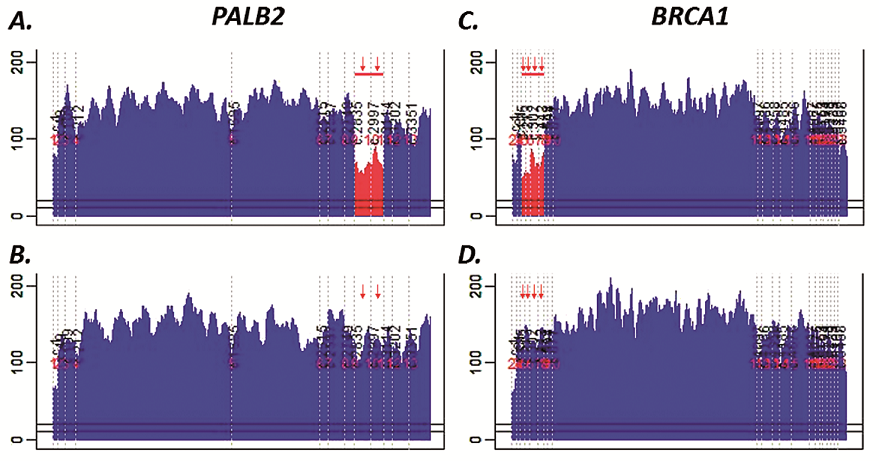
U vysoce rizikových osob vyšetřovaných na přítomnost nádorové predispozice ke vzniku dědičných forem karcinomu prsu lze nasazením panelového NGS do diagnostické praxe v současné době zvýšit záchyt patogenních či pravděpodobně patogenních variant o 10 % a především významně zkrátit dobu analýzy. Kromě toho současné technologie NGS umožňují při panelovém sekvenování získat poměrně spolehlivé kvantitativní informace o počtu osekvenovaných templátů (readů), což při dostatečném pokrytí (pro analýzu genetické predispozice - detekci heterozygotních variant, lze za dostatečné pokrytí považovat pokrytí v rozsahu ~35x.13 Vyšší pokrytí (100x) vyžaduje analýza přítomnosti intragenových přestaveb analyzovaných genů) umožňuje záchyt případných intragenových přestaveb (obr. 1). Panelové NGS je tak schopno nahradit nejen používané pre-screeningové metody pro detekci přítomnosti genetické varianty (např. HRM, dHPLC), samotné Sangerovo sekvenování pro charakterizaci genetické změny, ale i MLPA umožňující identifikaci intragenových přestaveb. Sangerovo sekvenování a MLPA jsou však nadále důležitými nástroji nezbytnými pro potvrzení nalezených patogenních variant.
Zatímco panelové NGS je po technické stránce již v současné chvíli vhodné pro klinické využití, největším omezením jeho rutinního použití je bioinformatická náročnost vyžadující expertní znalosti v oblasti zpracování rozsáhlých datových souborů a nejednoznačná klinicko-genetická interpretace charakterizovaných genotypů. Ta je nezbytná pro vyhodnocení, zda přítomnost (či chybění) nalezené varianty prokazatelně souvisí s výskytem onemocnění u postižené osoby a jejích příbuzných v rodině. Tento problémse týká nejen značné části kandidátních nádorových predispozičních genů, ale i řady variant (především missense variant a alterací v intronech a regulačních oblastech) v hlavních predispozičních genech (např. APC, TP53, BRCA1, MSH2), ve kterých je jinak význam vrozených mutací vedoucích ke zkrácení proteinového produktu nepochybný. Nemožnost jednoznačné klinické interpretace v současnosti vyplývá z doposud nedostatečného souboru vyšetřených osob (pacientů i kontrol), proměnlivé frekvence genetických variant v různých populacích ve světě a velmi nízké četnosti výskytu patogenních mutací v řadě genů. Velkým otazníkem je rovněž interpretace přítomnosti různých patogenních variant v nádorových predispozičních genech u jednoho nosiče.
Příspěvkem k překonání stávajících obtíží je snaha o integraci výsledků genetického testování na národní úrovni (v rámci projektu CZECANCA nebo Národního centra lékařské genomiky), ale i přesahujících aktivit na mezinárodní úrovni formou zapojení do mezinárodních konsorcií (např. ENIGMA, CIMBA, BCAC, COMPLEXO), která se podílejí na zpřesnění výpočtu rizika vyplývajícího z nosičství mutací v jednotlivých predispozičních genech, ale i na náročných funkčních in vitro analýzách identifikovaných genetických variant. Zásadním prostředkem, pomocí kterého lze překonat zmíněná omezení, je však používání panelového sekvenování (a v budoucnosti jeho rozšíření na exomové a genomové sekvenování) v onkogenetické diagnostice v ČR u dostatečně velkého souboru indikovaných osob (a osob kontrolního souboru). Pouze na základě analýzy zevrubných genetických charakteristik osob s vysokým rizikem nádorových onemocnění lze vybudovat hodnotnou databázi genotypů a jejich pečlivou korelací s fenotypovými projevy nádorových onemocnění zvýšit naše poznání genetické podstaty nádorového rizika a jeho variability u vysoce rizikových pacientů a jejich příbuzných. Výsledky tohoto úsilí bezpochyby umožní dále zvýšit klinickou použitelnost NGS v rutinní onkogenetické diagnostice a tím zlepšit prevenci nádorových onemocnění v rodinách s dědičnými nádorovými syndromy.